In [53]:
import time
import tensorflow as tf
config = tf.ConfigProto()
config.gpu_options.allow_growth = True
tf.enable_eager_execution(config=config)
import os
import glob
import numpy as np
from molmod.io import FCHKFile # https://molmod.github.io/molmod/reference/io.html#module-molmod.io.fchk
import multiprocessing
import seaborn as sns
import matplotlib
from matplotlib import pyplot as plt
from scipy.linalg import block_diag
import sys
sys.path.append("../../../")
import tfani
import tfani.aev as AEV
%matplotlib inline
In [3]:
H_fchk = '../HCON/h.fchk'
H_Atom = FCHKFile(H_fchk, ignore_errors=True)
C_fchk = '../HCON/c.fchk'
C_Atom = FCHKFile(C_fchk, ignore_errors=True)
O_fchk = '../HCON/o.fchk'
O_Atom = FCHKFile(O_fchk, ignore_errors=True)
N_fchk = '../HCON/n.fchk'
N_Atom = FCHKFile(N_fchk, ignore_errors=True)
HCNO=[H_Atom, C_Atom, N_Atom, O_Atom]
HCNO_list=['H_Atom', 'C_Atom', 'N_Atom', 'O_Atom']
HCNO_Atom_MO = {}
HCNO_Atom_MO.clear()
self_MO_energies = []
base_HCNO_densities = []
for i in range(len(HCNO)):
a = HCNO[i]
coordinates = a.fields['Current cartesian coordinates'] #Bohr
coordinates = coordinates * 0.529177249 #Angstrom
species = a.fields['Atomic numbers']
nuclear_charge = a.fields['Nuclear charges']
energy = a.fields['SCF Energy'] #Hartree
MO = a.fields['Alpha Orbital Energies']
MO_co = a.fields['Alpha MO coefficients']
density = np.reshape(MO_co, [int(np.sqrt(MO_co.shape[0])), -1])
base_HCNO_densities.append(density)
dipole = a.fields['Dipole Moment']
num_e = a.fields['Number of electrons']
print(num_e)
LUMO = MO[num_e//2]
HOMO = MO[num_e//2 - 1]
HCNO_Atom_MO[HCNO_list[i]] = MO
self_MO_energies.append(MO)
base_HCNO_densities[0].shape
Out[3]:
In [11]:
print(self_MO_energies[1])
print(self_MO_energies[2])
In [4]:
plt.figure(figsize=(16,2))
with sns.axes_style("white"):
ax = sns.heatmap(base_HCNO_densities[0], square=True, linewidths=.5, cmap="YlGnBu", annot=True, center=0.)
plt.xlabel('wavefunction', fontsize=14)
plt.ylabel('Coeffients', fontsize=14)
plt.show()

In [5]:
def plot_MO(ground_true, pred, i, hide_padding=True):
ground_true_data = ground_true[i]
pred_data = pred[i]
x = np.linspace(1, len(ground_true_data), num=len(ground_true_data))
diff = np.absolute(ground_true_data - pred_data)
diff_large_idx = np.squeeze(np.argwhere(diff>0.024))
diff_large_data = np.take(ground_true_data, diff_large_idx)
plt.figure(figsize=(18,10))
# plot diff > 0.04
plt.plot(diff_large_idx + 1, diff_large_data, label='diff > 0.024 hartree | 15.04 kcal/mol ({} points)'.format(len(diff_large_idx)),
marker='x', linestyle = 'None', markersize=10, markeredgewidth=1, color='magenta')
# plot ground ture
plt.plot(x, ground_true_data, label='ground true', marker='.', linestyle = '-', markersize=10,
markeredgewidth=1, color='r')
# plot pred
plt.plot(x, pred_data, label='prediction', marker='.', linestyle = '-', markersize=10,
markeredgewidth=1, color='g')
# title and label
plt.title('Using ANI to Predict Molecular Orbitals', fontsize=18)
plt.xlabel('Molecular Orbitals', fontsize=18)
plt.ylabel('MO Energy / hartree', fontsize=18)
plt.legend(frameon=False, fontsize=16)
plt.ylim(-20, 6)
plt.show()
In [6]:
plt.figure(figsize=(16,10))
with sns.axes_style("white"):
ax = sns.heatmap(base_HCNO_densities[2], square=True, linewidths=.5, cmap="YlGnBu", annot=True, center=0.)
plt.xlabel('wavefunction', fontsize=14)
plt.ylabel('Coeffients', fontsize=14)
plt.show()

In [51]:
# plt.figure(figsize=(16,10))
# with sns.axes_style("white"):
# ax = sns.heatmap(base_HCNO_densities[1]-base_HCNO_densities[2], square=True, linewidths=.5, cmap="YlGnBu", annot=True, center=0.)
# plt.xlabel('wavefunction', fontsize=14)
# plt.ylabel('Coeffients', fontsize=14)
# plt.show()
In [79]:
# C2NH5
tmp = FCHKFile('./gdb11_s03-0000024_0221.fchk', ignore_errors=True)
a = tmp
species = a.fields['Atomic numbers']
MO = a.fields['Alpha Orbital Energies']
MO_co = a.fields['Alpha MO coefficients']
density = np.reshape(MO_co, [int(np.sqrt(MO_co.shape[0])), -1])
num_e = a.fields['Number of electrons']
coordinates = a.fields['Current cartesian coordinates'] #Bohr
coordinates = coordinates * 0.529177249 #Angstrom
coordinates = np.reshape(coordinates, [-1, 3])
In [45]:
plt.figure(figsize=(16,10))
with sns.axes_style("white"):
ax = sns.heatmap(density, square=True, linewidths=.0, cmap="YlGnBu", annot=False, center=0., cbar=False)
plt.vlines(x=15, ymin=0, ymax=55, colors='darkgray')
plt.vlines(x=30, ymin=0, ymax=55, colors='darkgray')
plt.vlines(x=45, ymin=0, ymax=55, colors='darkgray')
plt.vlines(x=47, ymin=0, ymax=55, colors='darkgray')
plt.vlines(x=49, ymin=0, ymax=55, colors='darkgray')
plt.vlines(x=51, ymin=0, ymax=55, colors='darkgray')
plt.vlines(x=53, ymin=0, ymax=55, colors='darkgray')
for i in range(55):
plt.hlines(y=i, xmin=0, xmax=55)
plt.xlabel('wavefunction', fontsize=14)
plt.ylabel('Coeffients', fontsize=14)
plt.show()

In [16]:
pred = []
pred.append(self_MO_energies[1])
pred.append(self_MO_energies[1])
pred.append(self_MO_energies[2])
pred.append(self_MO_energies[0])
pred.append(self_MO_energies[0])
pred.append(self_MO_energies[0])
pred.append(self_MO_energies[0])
pred.append(self_MO_energies[0])
pred = np.concatenate(pred, axis=0)
In [17]:
plot_MO([MO], [pred], 0)

In [18]:
idx = np.argsort(pred)
sort_pred = np.take(pred, idx)
In [19]:
plot_MO([MO], [sort_pred], 0)

In [ ]:
In [33]:
NCCHHHHH = block_diag(base_HCNO_densities[1], base_HCNO_densities[1], base_HCNO_densities[2], base_HCNO_densities[0],
base_HCNO_densities[0], base_HCNO_densities[0], base_HCNO_densities[0], base_HCNO_densities[0])
plt.figure(figsize=(16,10))
with sns.axes_style("white"):
ax = sns.heatmap(NCCHHHHH, square=True, linewidths=.0, cmap="YlGnBu", annot=False, center=0.)
plt.hlines(y=15, xmin=0, xmax=30)
plt.vlines(x=15, ymin=0, ymax=30)
plt.hlines(y=30, xmin=15, xmax=45)
plt.vlines(x=30, ymin=15, ymax=45)
plt.hlines(y=45, xmin=30, xmax=55)
plt.vlines(x=45, ymin=30, ymax=55)
plt.xlabel('wavefunction', fontsize=14)
plt.ylabel('Coeffients', fontsize=14)
plt.show()

In [21]:
sort_NCCHHHHH = np.take(NCCHHHHH, idx, axis=0)
# sort_NCCHHHHH = np.take(np.transpose(NCCHHHHH), idx, axis=0)
# sort_NCCHHHHH = np.transpose(sort_NCCHHHHH)
In [44]:
plt.figure(figsize=(16,10))
with sns.axes_style("white"):
ax = sns.heatmap(sort_NCCHHHHH, square=True, linewidths=.0, cmap="YlGnBu", annot=False, center=0., cbar=False)
plt.vlines(x=15, ymin=0, ymax=55, colors='darkgray')
plt.vlines(x=30, ymin=0, ymax=55, colors='darkgray')
plt.vlines(x=45, ymin=0, ymax=55, colors='darkgray')
plt.vlines(x=47, ymin=0, ymax=55, colors='darkgray')
plt.vlines(x=49, ymin=0, ymax=55, colors='darkgray')
plt.vlines(x=51, ymin=0, ymax=55, colors='darkgray')
plt.vlines(x=53, ymin=0, ymax=55, colors='darkgray')
for i in range(55):
plt.hlines(y=i, xmin=0, xmax=55)
plt.xlabel('wavefunction', fontsize=14)
plt.ylabel('Coeffients', fontsize=14)
plt.show()

In [ ]:
In [70]:
# constants
constants = {
'Rcr': 5.2000e+00,
'Rca': 3.5000e+00,
'EtaR': [1.6000000e+01],
'ShfR': [9.0000000e-01,1.1687500e+00,1.4375000e+00,1.7062500e+00,1.9750000e+00,
2.2437500e+00,2.5125000e+00,2.7812500e+00,3.0500000e+00,3.3187500e+00,
3.5875000e+00,3.8562500e+00,4.1250000e+00,4.3937500e+00,4.6625000e+00,4.9312500e+00],
'Zeta': [3.2000000e+01],
'ShfZ': [1.9634954e-01,5.8904862e-01,9.8174770e-01,1.3744468e+00,1.7671459e+00,
2.1598449e+00,2.5525440e+00,2.9452431e+00],
'EtaA': [8.0000000e+00],
'ShfA': [9.0000000e-01,1.5500000e+00,2.2000000e+00,2.8500000e+00],
'num_species': 4}
In [81]:
_, aev = AEV.AEVComputer(**constants)(([tf.cast(species, tf.int64)], [tf.cast(coordinates, tf.float32)]))
aev = tf.squeeze(aev, axis=0)
In [67]:
print(species)
In [84]:
plt.figure(figsize=(60,30))
with sns.axes_style("white"):
ax = sns.heatmap(aev, square=True, linewidths=.0, cmap="YlGnBu", annot=False, center=0., cbar=False)
plt.xlabel('AEV', fontsize=14)
plt.ylabel('Atom', fontsize=14)
plt.show()

In [85]:
aev
Out[85]:
Think about how to improve AEV? ¶
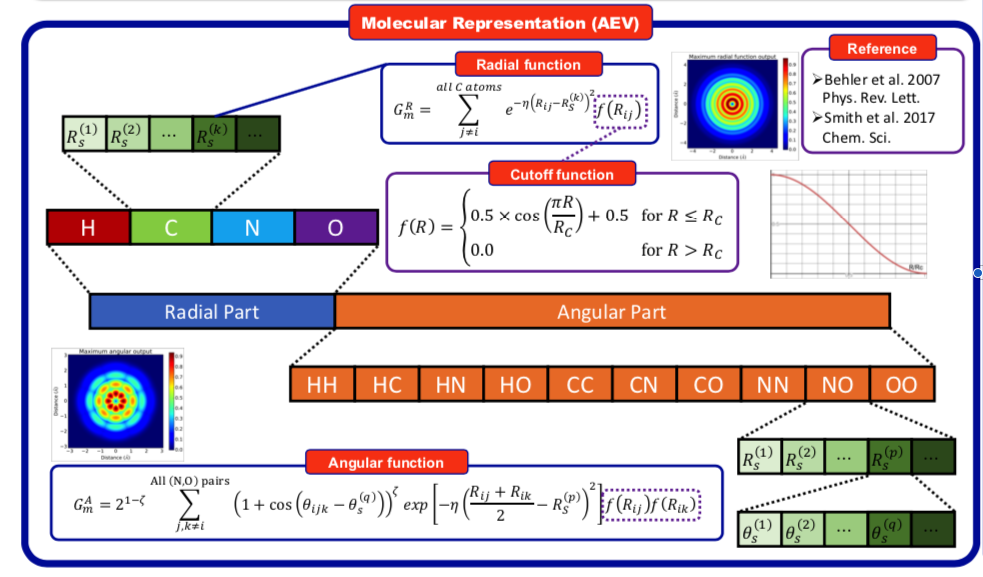
In [ ]: